
A new $1.5 million award from the National Institutes of Health will allow a University of Arkansas chemist to develop mathematical models to improve the reliability and efficiency of computer-aided drug design. The research could reduce the cost of drug discovery and lead to improvements in public health.

Feng Wang
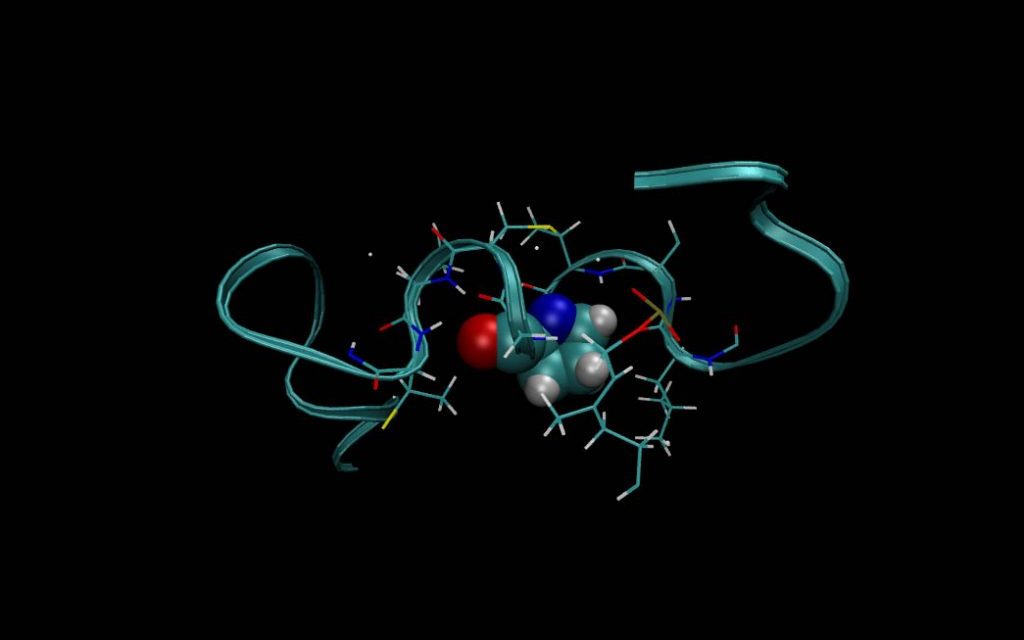
Computer-aided drug design is a critical component of drug discovery and the further development of more efficient, or targeted, pharmaceuticals. However, current models used in these designs have demonstrated limited accuracy in describing the interactions between drug molecules, their targets and their shared environment.
Feng Wang, associate professor of physical chemistry in the J. William Fulbright College of Arts and Sciences, has developed a new method for creating models that more accurately predict guest-host interactions and the binding affinities of proteins and ligands – molecules that attach to proteins to form a complex that regulates a biological function.
Wang’s method, called adaptive force matching, is an automated protocol that maps the complex landscape of molecular energy into simple mathematical forms.
“The significance of the proposed research is to enable predictive simulations of fundamental properties of drug candidates,” said Wang. “With adaptive force matching, we propose to shift the focus from creating a general-purpose force field to a rigorous protocol that will enable more efficient and accurate computational studies of the structure and function of drug candidates.”
One aim of the research is to create simulations to identify so-called nisin derivatives, the foundation for a class of drugs – generally referred to as lantibiotics – that hold great promise in addressing bacterial infections that are resistant to common antibiotics.
-30-


You must be logged in to post a comment.