Models Explain How Proteins Function at the Molecular Level
Mahmoud Moradi’s research could provide a framework for a vaccine targeting the activation of coronavirus spike proteins.

Chemist Mahmoud Moradi develops computational models that explain movement and behavior of proteins to determine how they function and how they attach to human cells and cause infection, which is a vital step in developing life-saving treatments and vaccines.
In late 2019 Moradi was studying hemagglutinin, a critical protein of the influenza virus that is a target for vaccine development, and had applied for a grant from the National Institutes of Health. Then, with the arrival of COVID-19 a few months later, Moradi, an assistant professor of chemistry and biochemistry at the University of Arkansas, shifted his research focus.
The shift was logical and relatively minor. Hemagglutinin is the influenza equivalent of the spike glycoproteins found on the novel coronavirus, which causes COVID-19.
“That NIH application for the hemagglutinin study happened months before we knew about COVID,” Moradi said. “For me, it functioned as the gateway into the topic.”
Trained as a bio-physicist, Moradi’s research lies at the confluence of biology, physics, chemistry, mathematics, statistics and computer science. He uses all of these disciplines to develop biomolecular simulations and computational theories that explain how proteins function at the molecular level. The theories improve geometric models to describe how proteins change their shape prior to binding to human cell receptors and how these changes affect virus behavior.
Breakthroughs Lead to 2020 Early Career Award
For years, theoreticians have been trying to devise mathematical shortcuts to accelerate computer simulations of protein behavior without compromising reliability. These shortcuts, called enhanced sampling techniques, are based on Euclidean geometry, which is embedded in conventional statistical mechanics.
Moradi improved enhanced sampling techniques by incorporating a general and more accurate geometry, known as Riemannian geometry, which allows the intrinsic protein space to be curved, somewhat similar mathematically to Einstein’s general relativity theory, where gravity curves, or warps, spacetime. This refinement enabled him to create even better simulations.

In early 2020 the National Science Foundation rewarded Moradi for this work, granting him a $650,000 Faculty Early Career Development award to continue his work modeling the function of proteins at the molecular level. This investigation, the NSF award said, will deepen our understanding of disease and improve drug design.
Simulating Spike Proteins to Fight COVID-19
Only weeks before Moradi received his Early Career award, COVID-19 surfaced in Wuhan, China. As the coronavirus spread quickly and led to the worst pandemic in more than a century, scientists rushed to respond to the problem. Epidemiologists and drug companies launched investigations to discover a vaccine.
Moradi joined the effort with a strong foundation from his previous research on modeling protein form and function. Only months before, he had applied for the NIH grant to study the protein hemagglutinin — the functional influenza equivalent of the now famous spike glycoproteins proteins found on the novel coronavirus, COVID-19.
As part of the COVID-19 High Performance Computing Consortium, a collaboration of government, industry and academic partners focused on computing resources for COVID-19 research, Moradi was granted computing time on Frontera, an NSF-sponsored supercomputer at the University of Texas at Austin and the largest supercomputer on any university campus.
Benefitting from several recent, high-resolution, 3D static models, which he used as the initial structures, Moradi harnessed Frontera’s power and processing speed to develop 3D simulations of the molecular dynamics of coronavirus spike glycoproteins.
His goal was to map novel coronavirus spike proteins and show how they undergo structural and conformational changes prior to binding to host human cell receptors.
“As with other viruses, a crucial step in the coronavirus infection process is viral entry,” Moradi said. “If we can develop dynamic, three-dimensional visualizations of cell structures and the behavior of these proteins – in other words, animations or movies rather than static pictures – then we can understand entry better.”
What they know so far isn’t comforting.
As a starting point, Moradi and colleagues simulated and compared SARS-CoV-2, the virus that causes COVID-19, to the earlier coronavirus, the one that caused the 2003 epidemic, because the spike proteins of both attach to the same human cell receptor, called angiotensin-converting enzyme 2. Previous experiments had determined that static models of the spike proteins of both viruses did not indicate any major differences in shape.
But the dynamic simulations Moradi developed told a different story. Showing both viruses in active and inactive states, and during the activation and deactivation process, the simulations provided compelling evidence that the two otherwise homologous proteins have drastically different dynamics.
In addition to being significantly more active than the spike proteins of the earlier coronavirus, SARS-CoV-2 — COVID-19 — spike proteins were much more capable of attaching to human cells and thus infecting people. In the researchers’ simulations, SARS-CoV-1 spike proteins became inactivate within a few microseconds, while the SARS-CoV-2 spike proteins remained active for comparatively longer time.
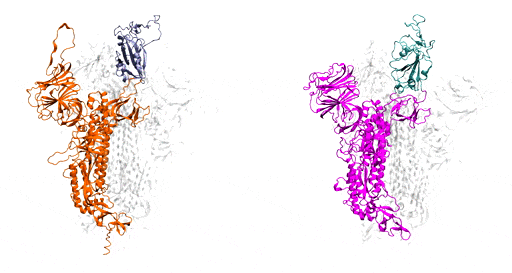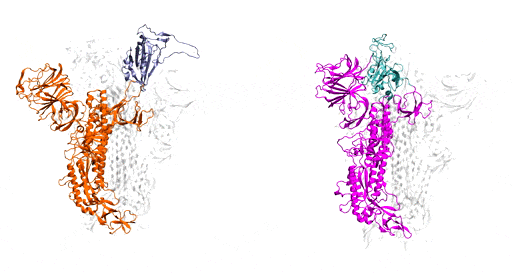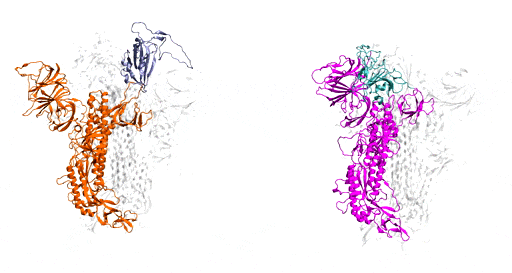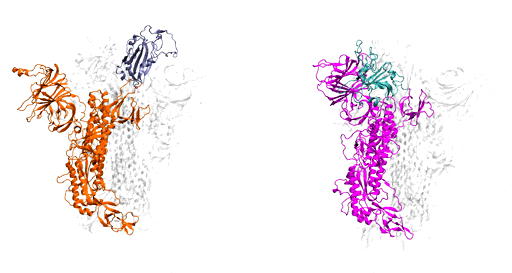
These dynamic models are important, Moradi said, because they provide a framework for a structure-based design of a reliable vaccine and possibly therapeutics that target the activation of the spike protein rather than its binding to human cells.
In other words, drugs would target the coronavirus before it approaches the human cell and block the activation of the virus.
“Relying only on the experimentally available, static models of SARS-CoV-2 spike proteins is misleading,” Moradi said. “We can clearly see in our simulations that these static models are incomplete.”
Shape and Motion
“We know proteins are not static objects. Understanding their conformational dynamics is a necessary step in deciphering the molecular mechanisms underlying their function,” Moradi said.
In 2017, Moradi articulated a new theory for explaining how the movement and shape of proteins and other biomolecules is a key element of their function. In a study published in The Journal of Physical Chemistry Letters, he and colleague Ashkan Fakharzadeh, a graduate student at North Carolina State University, argued that “conformational dynamics” — movement and change of shape and structure — determine the function of proteins and biomolecules.
The study found that researchers had historically focused solely on content, composition and shape of biomolecules. Because of this, researchers weren’t seeing the complete picture, Moradi said. His models fill in important gaps in knowledge by showing how molecules move and change their shape to conform and then bind to cell receptors.
“We were motivated to do this because conventional theories of protein dynamics ignored the curved nature of the configurational space of biomolecules,” Moradi said. “So we developed a kind of innovative formalism that relies on a geometric theory, traditionally used in general relativity and similar fields, to modify theories of protein dynamics.”
New Methods and Workhorse Molecules
Proteins are known as the workhorse molecules of cells. They are responsible for nearly every task in living systems. Some proteins live inside cells, and some reside on cell membranes, the outer layer of lipids that separates the cell from its external environment.
Membrane proteins are critically important because they regulate the exchange of information and materials between the cell and its environment.
This is a vital task for the survival and normal function of the cell. Any disorder in protein function can result in disease. Researchers focus on how membrane cells function to gain a deeper understanding of the molecular basis of disease. This knowledge then establishes a rational design for drug development.
For decades, researchers have relied on an experimental method called X-ray crystallography to study protein function. As the primary tool for determining the shape and structure of proteins, X-ray crystallography has been essential for the design of drugs that manipulate protein function – basically tricking them into behaving in an orderly fashion, so the cell will function normally, and diseases and disorders will not develop.
However, when applied to cell membrane proteins, X-ray crystallography has limits.
In 2019, Moradi and Thomas Harkey, an undergraduate student at the time and now a medical student at the University of Arkansas for Medical Sciences, published a study in Scientific Reports showing that X-ray crystallography can provide inaccurate information about proteins found on cell membranes and thus could be leading to poor and inefficient drug design.
This is especially problematic, Moradi emphasized, because roughly two-thirds of all drugs, including those used for chemotherapy, target proteins on cell membranes.
Moradi and Harkey provided evidence of the potential for inaccuracy by focusing on a membrane protein called YidC2, which, according to X-ray crystallography, has an unresolved cytoplasmic loop in its molecular structure. Cytoplasmic loops are known to have functional significance in membrane proteins. With use of a supercomputer at the Arkansas High Performance Computing Center, Moradi and Harkey ran continuous, microsecond-level computations simulating the molecular dynamics of YidC2.
Their simulations demonstrated that YidC2’s cytoplasmic loop stabilized the entire protein, particularly the C1 region, a potentially important area for drug design. This meant that unresolved cytoplasmic loops within membrane proteins could be important for their stabilization, despite their apparent lack of molecular structure.
“Typically, with X-ray crystallography, if part of a protein is not resolved, it is interpreted as lacking a particular structure, or order,” Moradi said. “Our research showed that for membrane proteins and particularly parts of the protein that interact with the cell membrane, this interpretation is not accurate and could be misleading.”
The study showed that computational chemistry and supercomputing technology can be used to accurately model membrane proteins in an environment that mimics their actual, physiological environment.
NIH Award
In August of 2020, the NIH informed Moradi that he would receive $422,578 for the influenza hemagglutinin study that sparked his recent work on COVID-19.
Moradi and researchers in his Biomolecular Simulations Group will focus on developing simulations of the pH-triggered activation mechanism of hemagglutinin. Previous research has shown that the activation of the hemagglutinin protein is caused by a change in the pH of its environment, a process that facilitates fusion between viral membranes and those on human cells. Researchers think that if this pH-triggered activation can be inhibited, it could provide a new framework for developing universal vaccines and therapeutics for influenza.
In addition to the NIH grant and NSF Early Career Award he received this year, Moradi’s research benefits from use of several large supercomputers sponsored by the National Science Foundation. He recently received computing allocation equal to $3.3 million from the Extreme Science and Engineering Discovery Environment program, the NSF’s virtual system for helping scientists share computing resources, data and expertise.
Learn more about Moradi’s work on protein modeling in a recent episode of Short Talks From the Hill, the research podcast of the University of Arkansas.